
Peptide catalysts

Little is known about the principles that govern the catalytic activity of short peptides at the sequence level. It appears that the residues that make up the sequence but also the order in which they appear are important when designing catalytic peptides. Many approaches tend to mimic the catalytic triad of a desired enzyme such as Ser-His-Asp. Remarkably, even very short peptides such as seryl-histidine (Ser-His) were shown to catalyze a number of hydrolytic reactions in water. Similarly to the serine protease chymotrypsin, which cleaves proteins and carboxyl esters, Ser-His dipeptide was found to cleave the p-NPA ester.
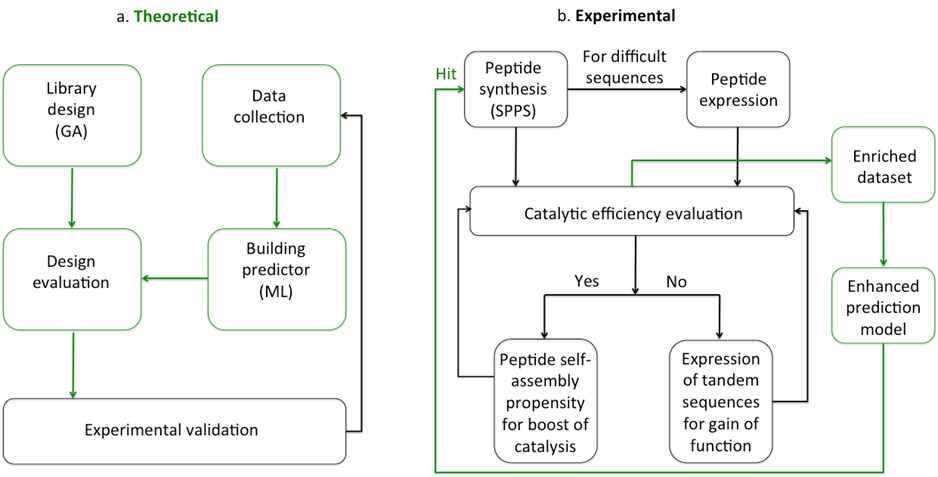
Even though several peptides have been shown catalytic activity as part of various supramolecular structures, the principles that govern their catalytic propensity at the sequence level have not been explored yet. It is our goal to understand how a particular peptide sequence acquires catalytic activity and how can sequences evolve to reach function. We want to gain information on monomers (combinations of amino acids) through a machine learning strategy and establish how to design better sequences in terms of their catalytic efficiency. Next, we will seek to create complexity through SA propensity to boost their activity.
Machine learning for peptide design
Machine learning (ML) is a niche in data science and a subset of artificial intelligence (AI) that computationally searches for patterns or regularities in data, enabling us to understand a phenomenon or to make predictions in the near future. By learning from data, ML uses the theory of statistics to build a
mathematical model and to make up for the lack of knowledge. The main application of ML is to help us find solutions to problems for which the amount and dimensionality of data is too overwhelming to comprehend. The analysis of chemical search space is such a problem domain.
Chemistry costs both time and physical resources, resulting in a limit on the number of experiments and the speed at which they can be performed. To date, artificial intelligence has been applied to a variety of chemical problems to maximize the chance of successful and rapid solving of complex issues. ML and evolutionary algorithms are being increasingly proposed to address various challenges of peptide chemistry related to the design and identification of peptides with a desired function. We reported on a multi-objective evolutionary approach for the design of mass and sequence diversity-oriented random peptide libraries.
Prediction models have been adapted for discovery of antimicrobial, antiviral and anticancer peptides. However, peptides with catalytic activity have not been searched before, using this strategy. Inspired by the recent advances in the field of antimicrobial peptides made by the combination of ML and experimental validation, we aim to determine whether it is possible to predict catalytically active peptides.
Optimization based on evolutionary algorithms
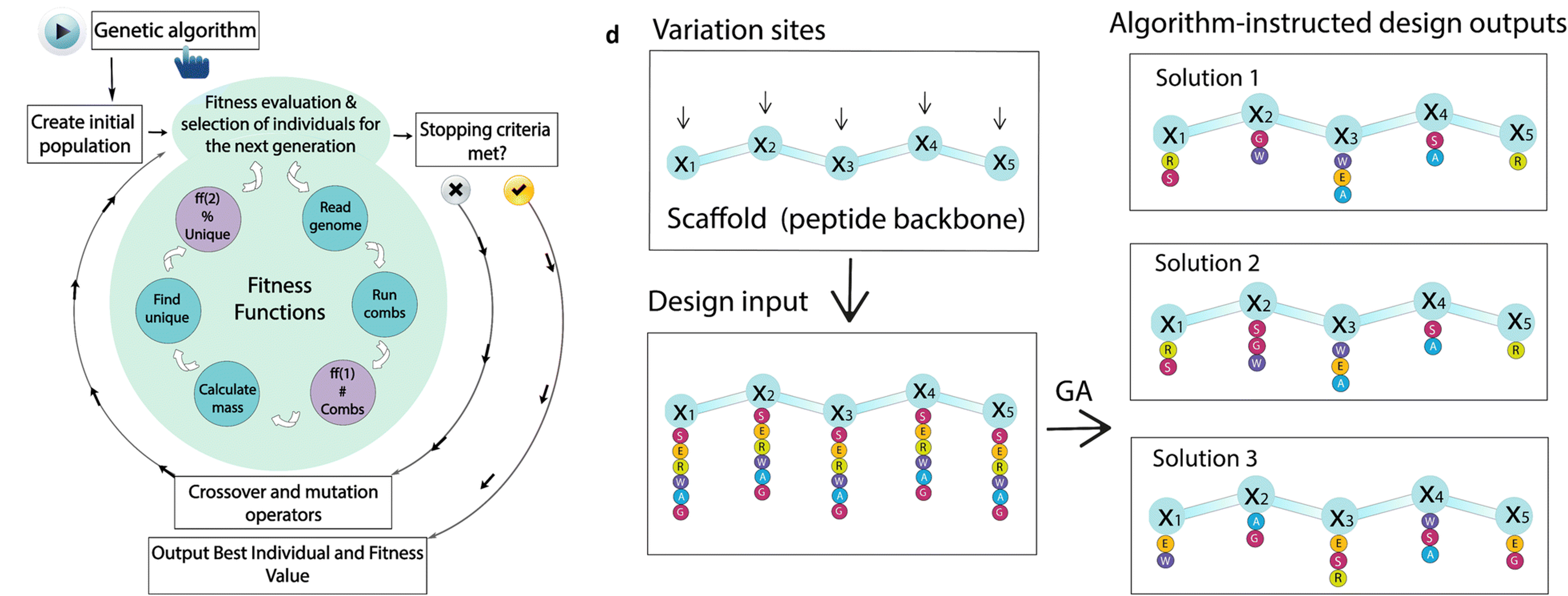
The 20 gene-encoded amino acids, together with a variety of non-natural ones, constitute a versatile toolbox to obtain peptides with high chemical and structural diversity. The number of possible permutations grows exponentially with the peptide length, depending also on the number of amino acids per position. If all the 20 proteinogenic amino acids are used in r positions, there are 20 r possible permutations. Due to the huge sequence space available even for short sequences it is challenging to establish meaningful relationships between sequence (primary structure) and function. Therefore, we need to rely on the use of artificial intelligence to help us explore this large space.